
 科普中國公眾號
科普中國公眾號
 科普中國微博
科普中國微博

 幫助
幫助
 HyperAI超神經
HyperAI超神經 
自 2004 年開源發布以來,LAMMPS 在材料建模領域得到了廣泛應用,其全稱為 Large-scale Atomic/Molecular Massively Parallel Simulator,由美國 Sandia 國家實驗室開發。
LAMMPS 可用于固態材料(金屬、半導體)和生物分子、聚合物等多種材料的建模,能夠為不同材料提供多種粒子相互作用模型
更重要的是,LAMMPS 既可在單個處理器上運行,也可使用消息傳遞技術和模擬域的空間分解技術并行運行。其代碼設計易于修改或擴展新功能,許多模型都有在 CPU、GPU 和 Intel Xeon Phis 上提供加速性能的版本。
截至 2022 年,已有數百人為 LAMMPS 貢獻了新的功能,其代碼行數也從 2004 年的 5 萬行增長到了 2022 年的 100 萬行。
為了方便大家體驗這個經典的分子動力學模擬軟件,HyperAI超神經官網的教程板塊現已上線了「LAMMPS 入門教程:npt 控溫估計 FCC Cu 熔點」,使用 CPU 版本的 LAMMPS 即可運行。
效果示例:

通過本教程的學習,您將能夠:
* 理解 npt 控溫操作流程
* 使用 dump 和 fix 指令將數據預處理
Demo 運行
啟動容器
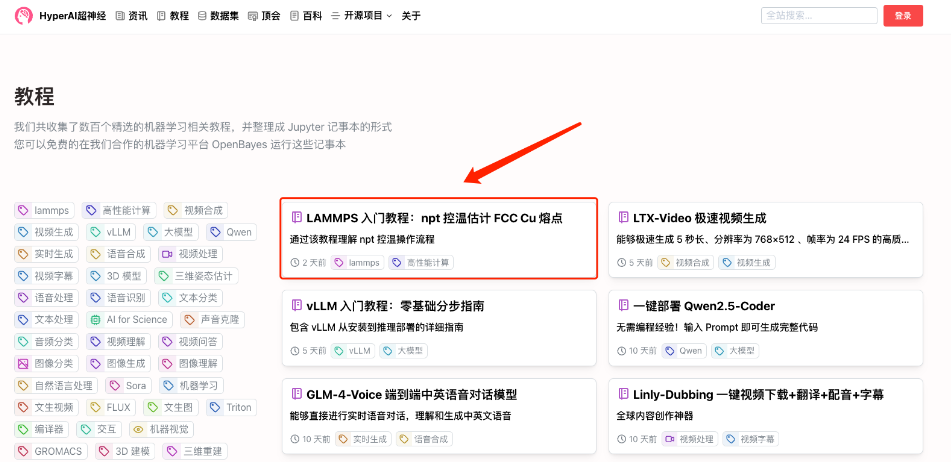
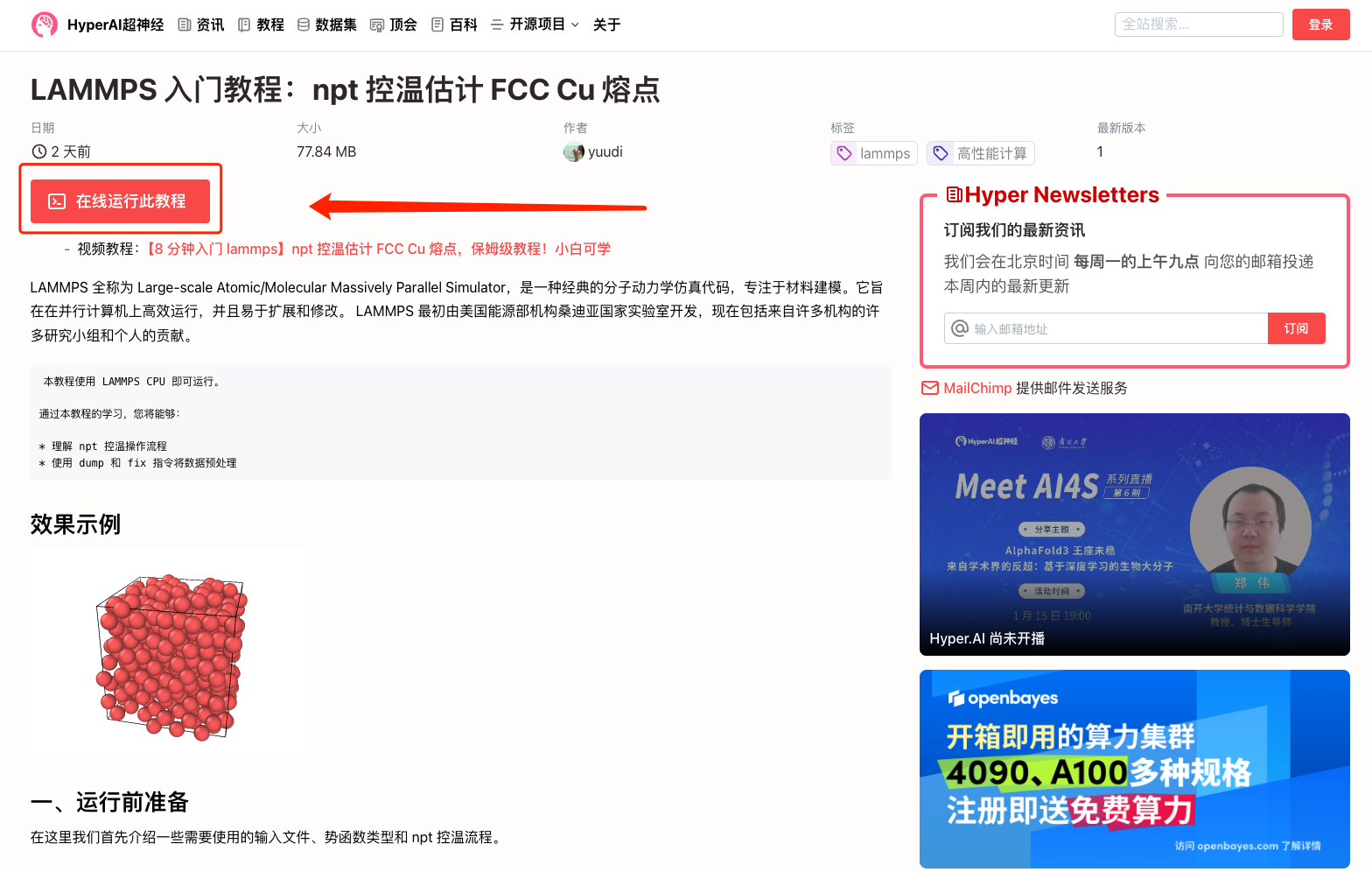
1. 登錄 hyper.ai,在「教程」頁面,選擇「LAMMPS 入門教程:npt 控溫估計 FCC Cu 熔點」,點擊「在線運行此教程」。
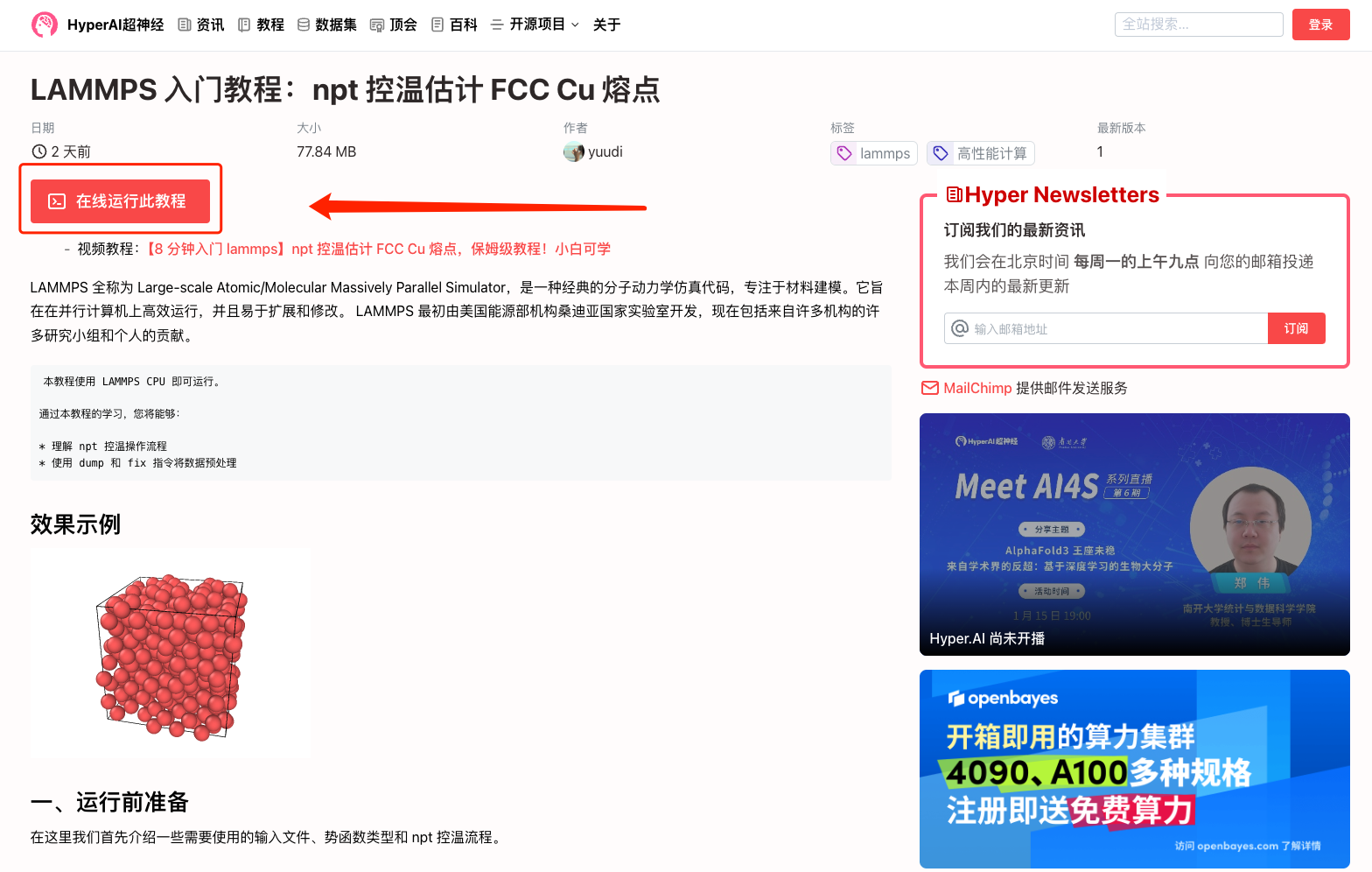
2. 頁面跳轉后,點擊右上角「克隆」,將該教程克隆至自己的容器中。
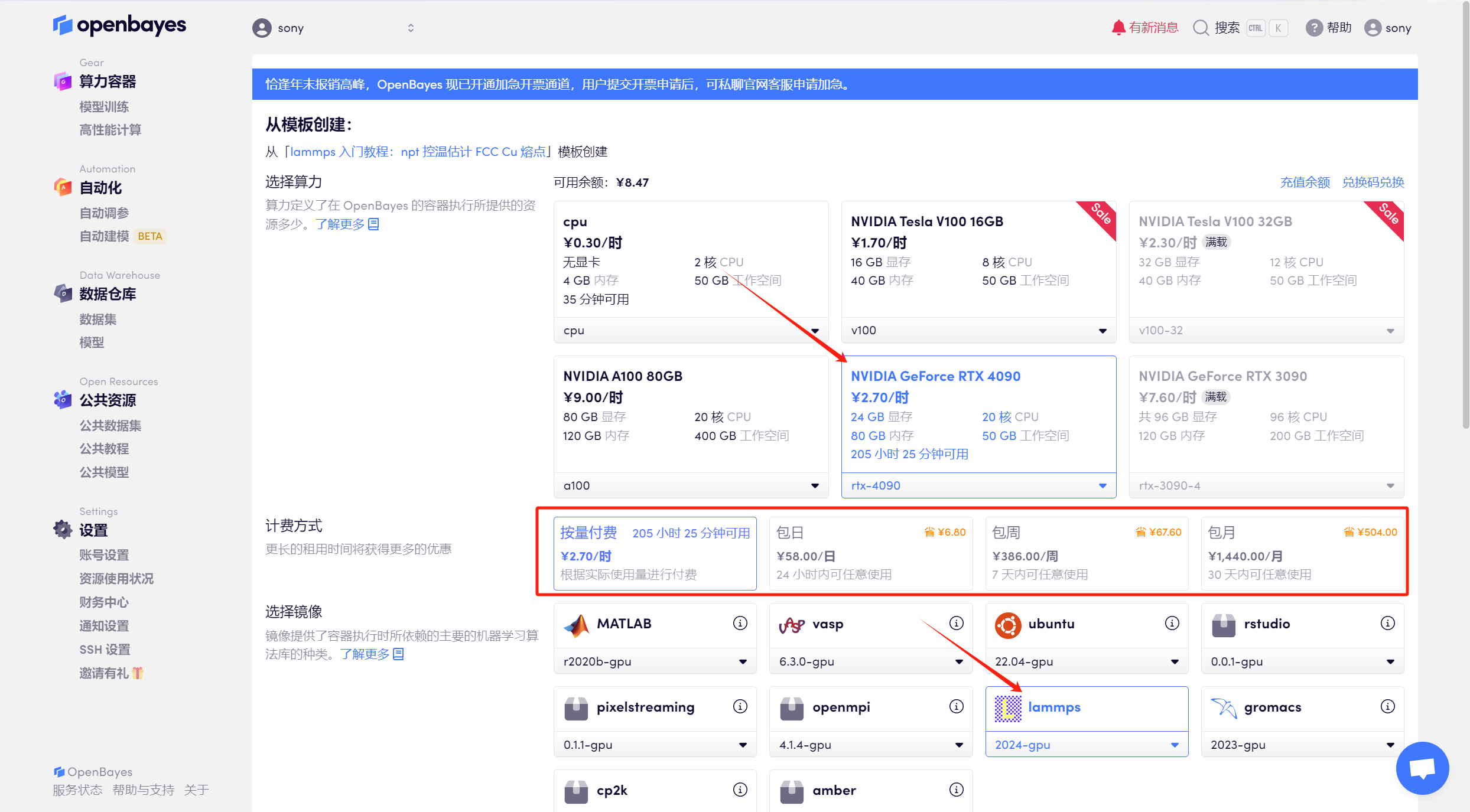
3. 選擇「NVIDIA RTX 4090」算力,按照自己需求選擇「按量付費」或「包日/周/月」,鏡像選擇「lammps」,最后點擊「繼續執行」。
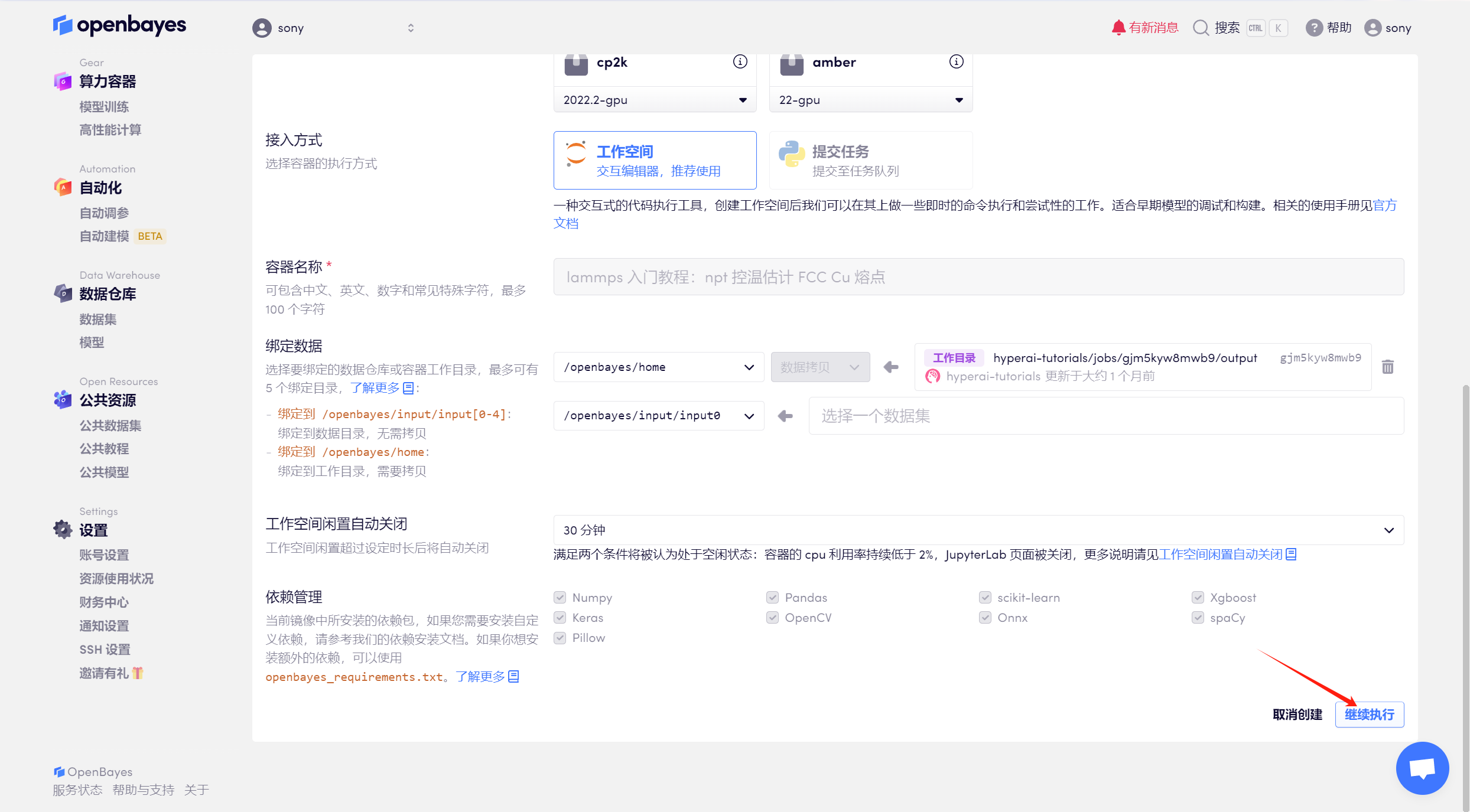
4. 等待模型分配好資源,狀態變為「運行中」后,點擊「打開工作空間」。
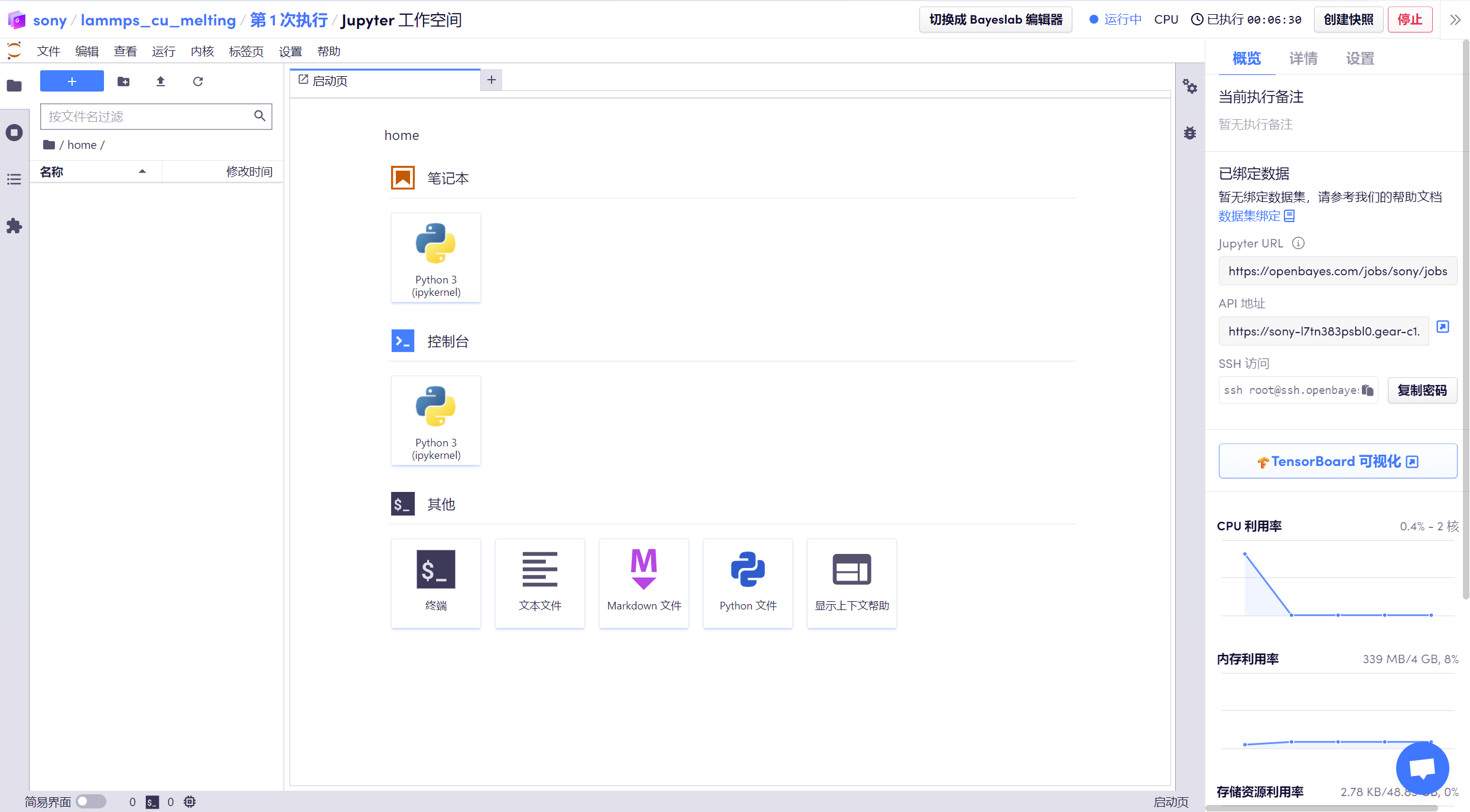
運行步驟
1. 進入到「工作空間」后,可以看到已經準備好的「melt_u3.zip」壓縮包,該包內含已經輸入好的相關命令,如系統定義、讀取銅結構、使用銅的 eam 勢函數等。
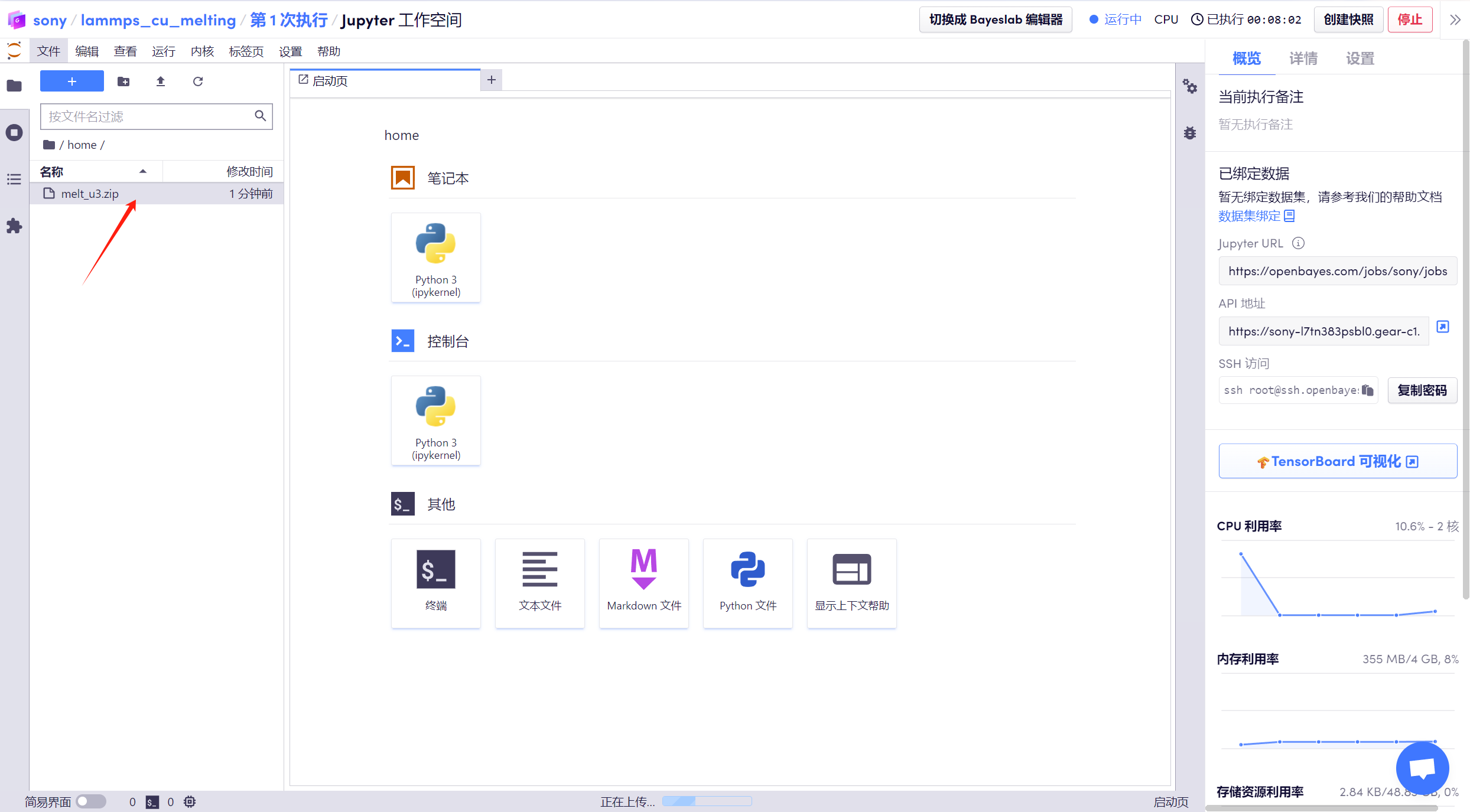
2. 打開「終端」,輸入「cd melt_u3」進入解壓目錄,使用「ls」命令查看文件。
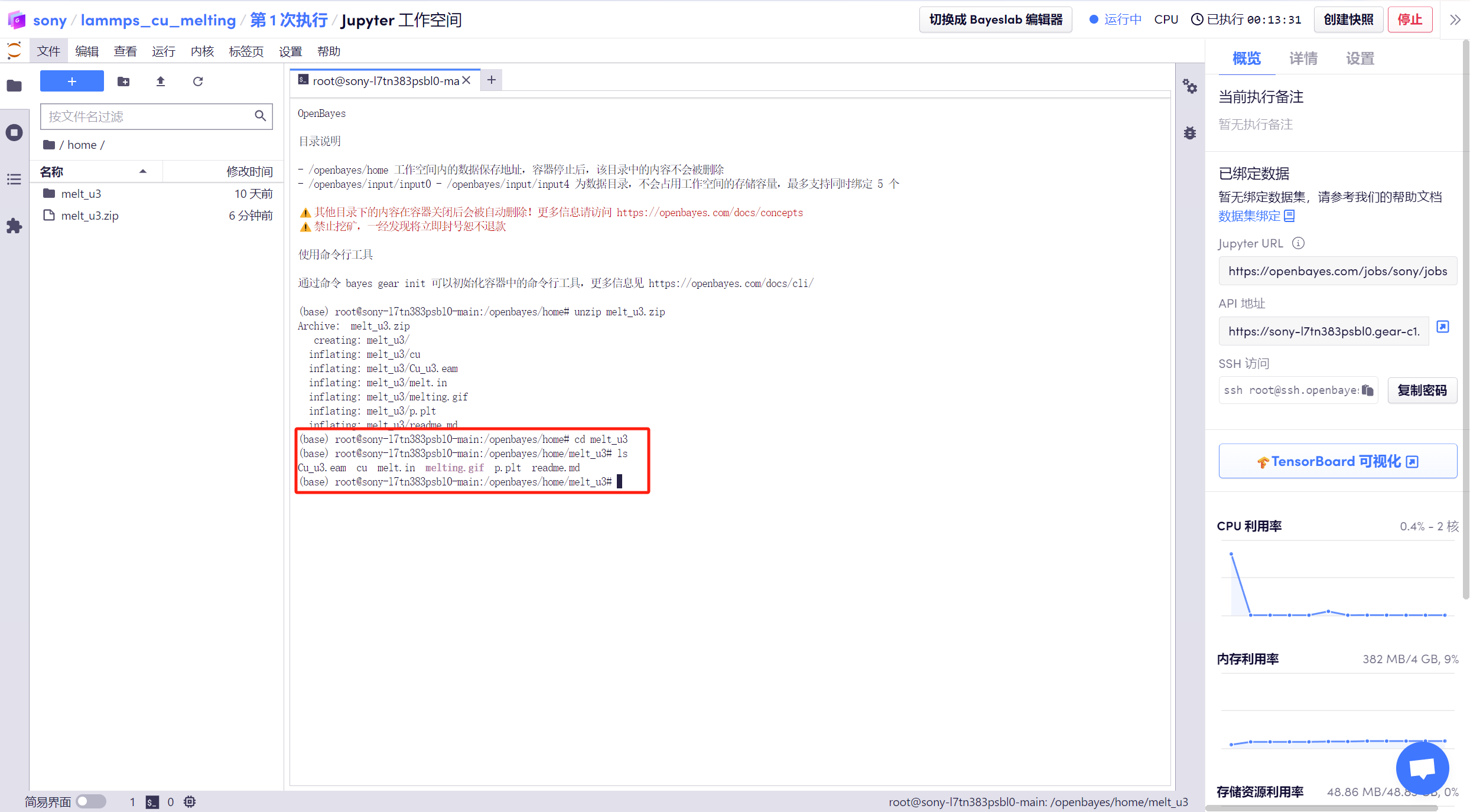
3. 輸入「mpirun -np 2 lmp < melt.in | tee out」,運行 lammps ,整個過程大約需要 5-10 分鐘。
運行完畢后可在文件夾中得到「t_v.txt」等輸出文件,其中溫度體積數據都輸入到了「t_v.txt」文件中,后面我們需要通過畫圖工具將數據可視化。
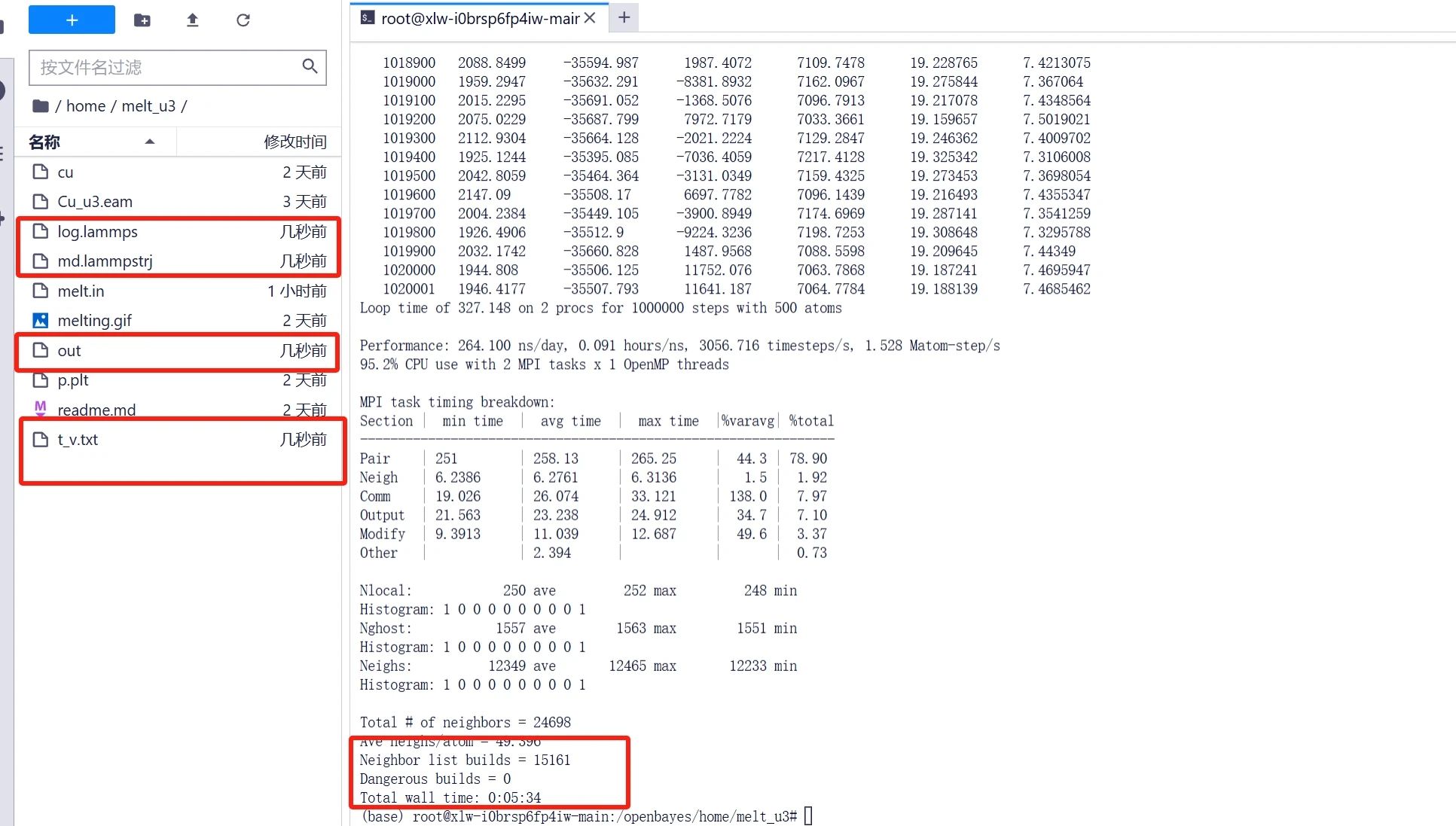
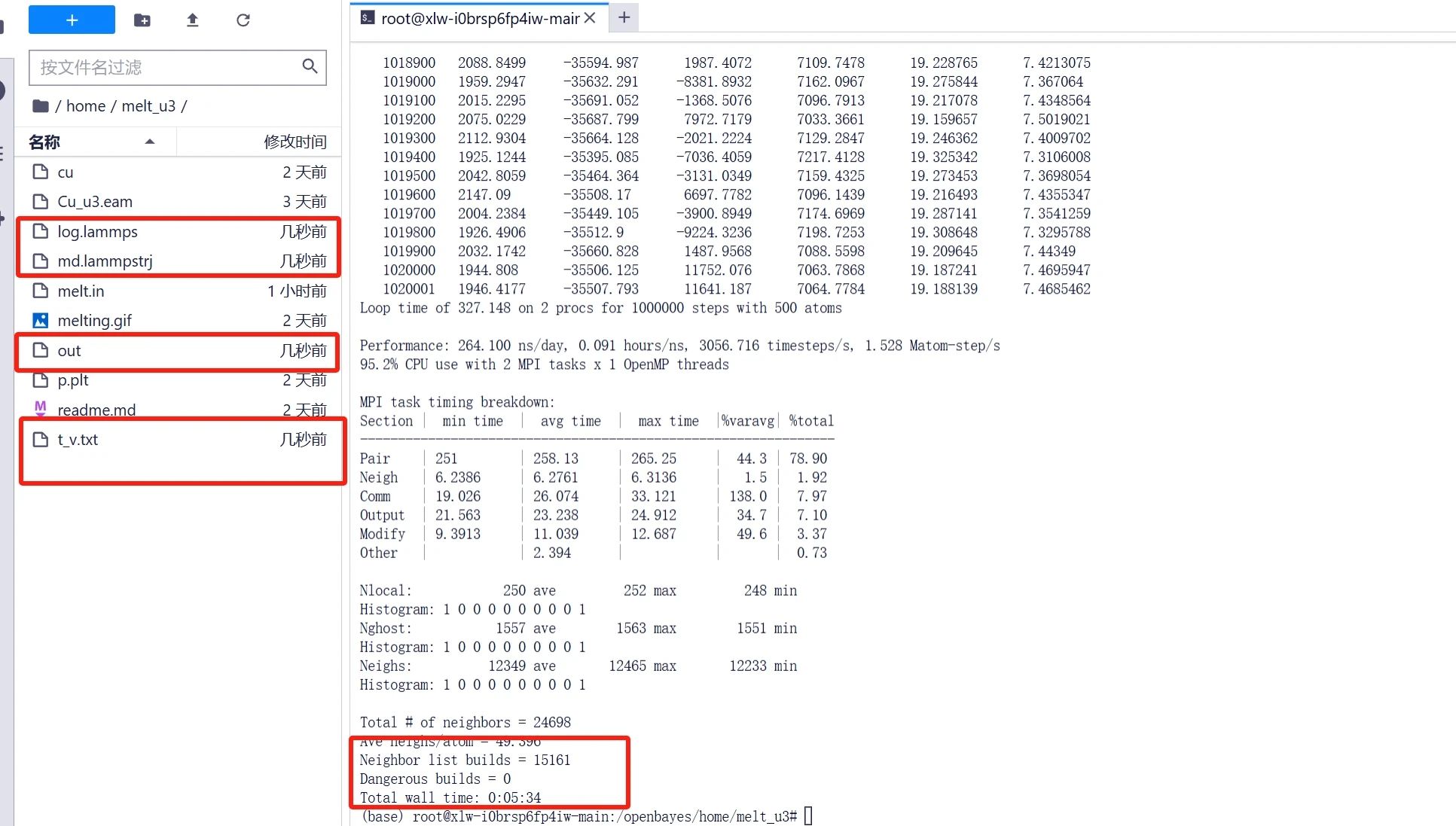
數據處理
1. 等待模型運行完成后,輸入「apt-get update --fix-missing」更新 apt 源,更新好之后輸入「apt install gnuplot」安裝 gnuplot(畫圖工具),并輸入「y」回車確認。

2. 用剛安裝好的畫圖工具將數據可視化。
運行腳本已經寫好,直接運行命令「gnuplot p.plt」可得到 t-v 圖即溫度體積階躍曲線圖,可以看到階躍點、也就是熔點為 1600k。
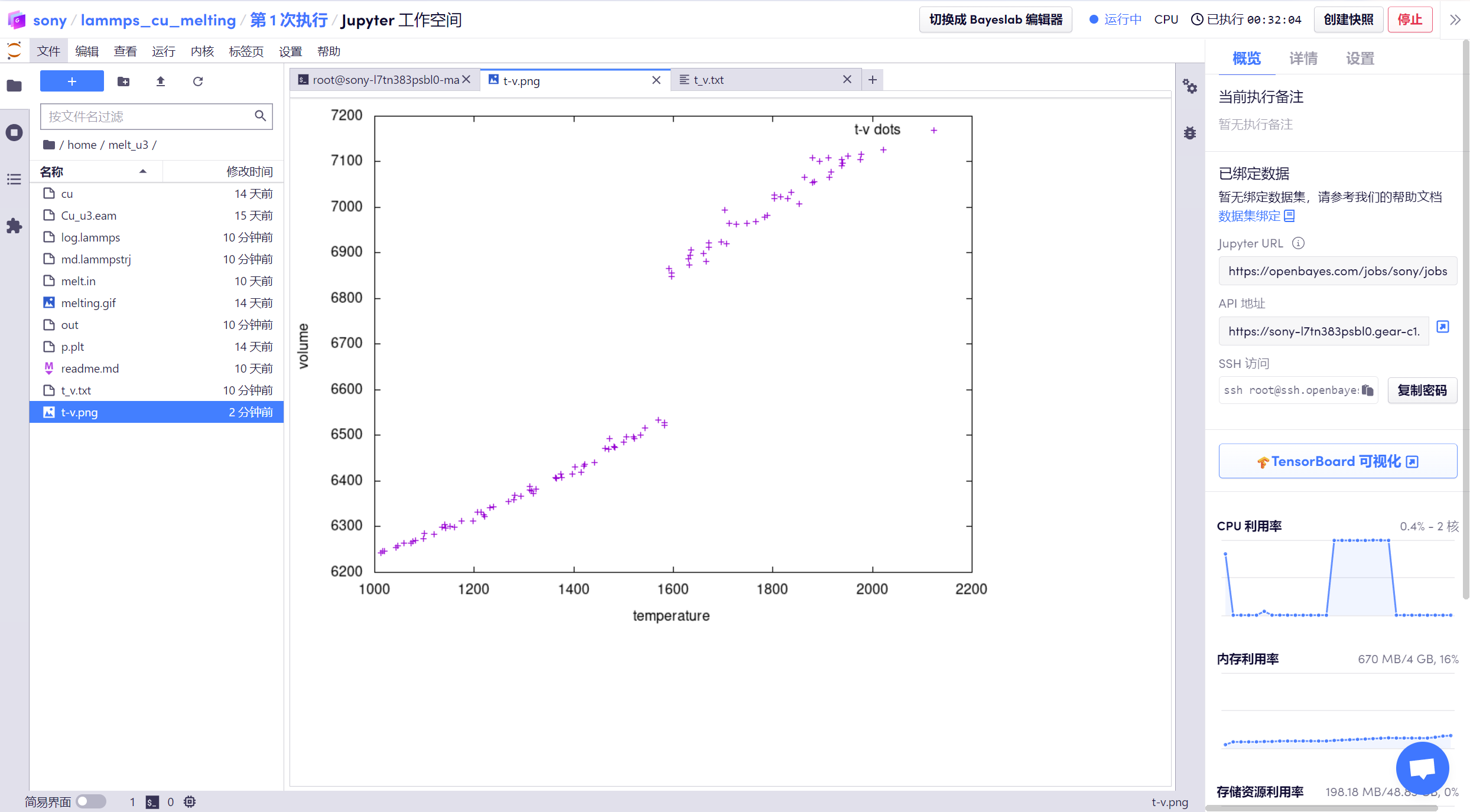
3. 然后將其原子軌跡文件「md.lammpstrj」下載到本地。
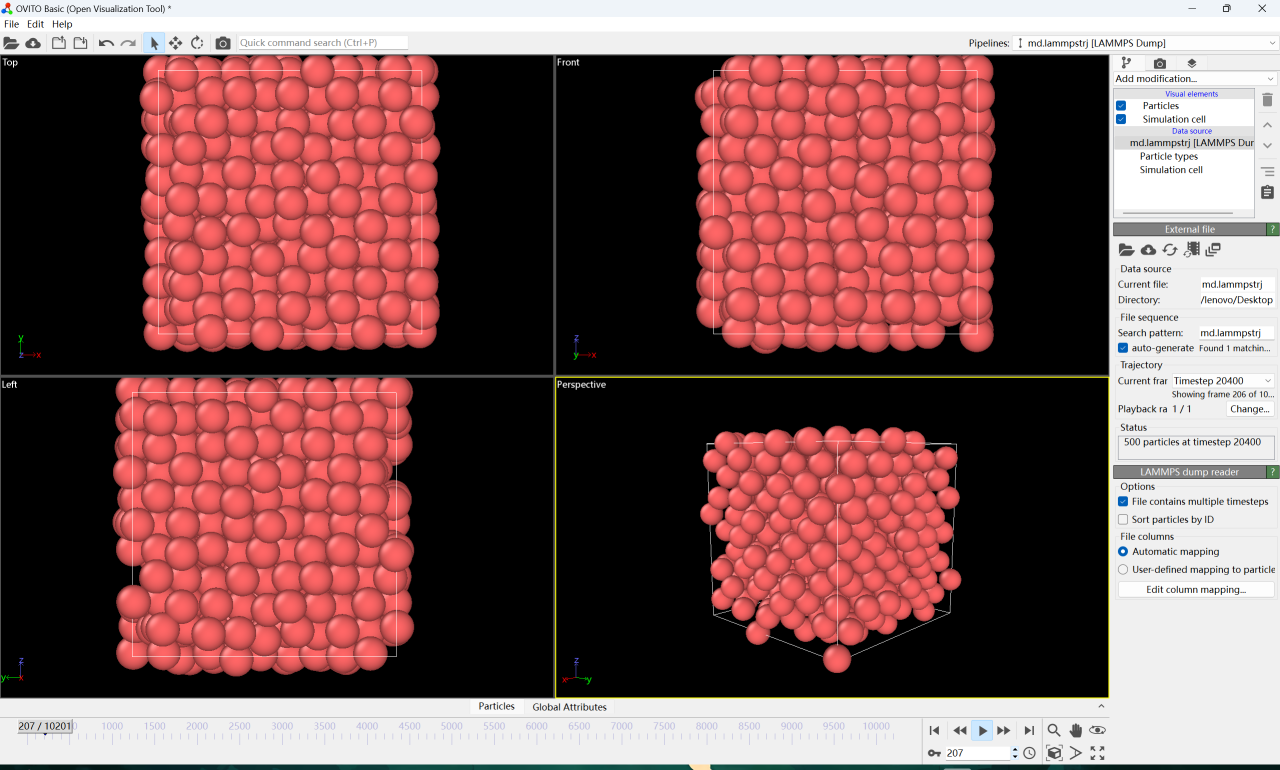
4. 將下載的文件放入 OVITO 中打開,點擊播放即可看到銅溶解過程中每個銅原子的運行軌跡。
我們建立了「Stable Diffusion 教程交流群」,歡迎小伙伴們入群探討各類技術問題、分享應用效果~
添加神經星星微信(微信號:Hyperai01),備注「SD 教程交流群」,即可加入群聊。
來源: HyperAI超神經
