
 科普中國公眾號
科普中國公眾號
 科普中國微博
科普中國微博

 幫助
幫助
 HyperAI超神經
HyperAI超神經 
作者:彬彬
編輯:李寶珠,三羊
浙江大學與之江實驗室研究團隊提出了一種基于蛋白質口袋 (protein pocket) 的 3D 分子生成模型——ResGen,與以往最優技術相比,速度提升 8 倍,成功地生成了具有更低結合能和更高多樣性的類藥物分子。
過去,創新藥物的發現往往依賴于古早配方或實驗中的偶然事件,例如青霉素。多年來,分子生物學和計算化學的進步,使藥物設計模式實現了從盲目篩選到合理設計的轉變。
盡管如此,藥物研發設計仍然是一個多環節流程,鏈路長且成本高昂,每一個環節的效率提高都有巨大價值。近年來,隨著 AI、大數據等技術的廣泛應用,AI 輔助藥物設計也在一次次的實驗中愈發成熟,AI 正在藥物研發的多個環節進行著提效增質的升級改革。
其中,高質量的分子生成模型可以有效提升先導化合物發現的效率。目前,大多分子生成工作都采用了基于配體的方法 (LBMG),然而該方法存在諸多局限性,例如無法考慮分子與靶標之間相互作用模式等。因此研究者們越來越關注基于結構的分子生成 (SBMG,structure-based molecular generative) 的方法,即基于靶標結構進行相應的分子生成。
浙江大學侯廷軍教授、謝昌諭教授和之江實驗室陳廣勇聯合團隊,提出了一種以蛋白質口袋為條件的 3D 分子生成模型——ResGen。該模型采用并行多尺度建模策略,可以捕捉到蛋白靶點與配體間更高層次的相互作用,并實現更高的計算效率。
分子生成過程被表述為全局自回歸和原子自回歸,以更好地考慮蛋白質口袋的幾何形狀。研究結果表明,與現有最先進方法相比,ResGen 生成的分子具有更合理的化學結構,并擁有更好的靶點親和能力。

獲取論文:
https://www.nature.com/articles/s42256-023-00712-7
公眾號后臺回復「3D 分子生成」獲取完整 PDF
數據集:訓練集與測試集間的序列相似性小于 40%
該研究使用的訓練數據集是 CrossDock2020,該數據集用于蛋白質-小分子相互作用研究,特別是用于評估分子對蛋白質口袋的結合能力。
該數據集的初始數據包含超過 2,200 萬個蛋白質-小分子配對 (protein–ligand pairs),為確保訓練集與測試集之間的序列相似性小于 40%,研究人員經過篩選,得到了約 10 萬個蛋白質-小分子配對,測試集中包含了 100 個蛋白質口袋。
數據集鏈接:
https://1lh.cc/DjuQrx
ResGen 模型:兩個分層自回歸
ResGen 模型將以蛋白質口袋感知為條件的分子生成問題,表述為兩個尺度的自回歸問題,即全局尺度和原子組件尺度。其中,全局自回歸 (global autoregression) 是指,ResGen 所生成的每個原子,都是基于之前步驟中生成的分子片段和蛋白質口袋結構;原子自回歸 (atomic autoregression) 依次產生新添加的原子坐標和拓撲。
ResGen 可以將完整的分子生成過程分解為分步采樣,從而以自回歸式方式實現整個分子的生成。此外,為了更好地捕獲更高層次的相互作用和降低計算成本,研究團隊在這個三維條件生成問題中引入了并行多尺度建模技術。

ResGen 框架示意圖
* 圖 A 示意:在分子生成的過程中,逐步地確認生長點,添加原子(全局自回歸),確認原子的位置,然后添加邊(原子自回歸)。
* 圖 B 示意:口袋和參考分子被表示成原子特征 (vector) 和原子坐標 (scalar)。
* 圖 E 示意:分子生成過程。i 中的灰色點云代表新生成的原子,具有位置信息;ii 中的綠色點云,是新生成的原子,補充了原子類型紅色圓圈表示每一步的焦點原子( focal atom,生長點),而數字是每個原子成為生長點的概率。
效果驗證:優于當前最優模型
一直以來,對于基于蛋白質口袋的 3D 分子生成模型有 2 個廣泛應用的檢驗指標——模型是否學習了配體在不同蛋白質口袋中的特征拓撲分布(即靶點的分子圖分布),以及口袋內配體的幾何分布(即原子位置和構象的合理性)。
對此,研究團隊對 ResGen 和現有的最先進模型進行了系列評估。
對于第一條檢驗指標,團隊評估了針對測試集中的靶點和真實的治療靶點設計生成的分子的結合能 (binding energies) 和類藥性 (drug-like properties)。
對于第二條檢驗指標,團隊設計了構象合理性實驗,并分析了蛋白與小分子之間的相互作用模式。
在測試集上生成分子:評估模型泛化能力
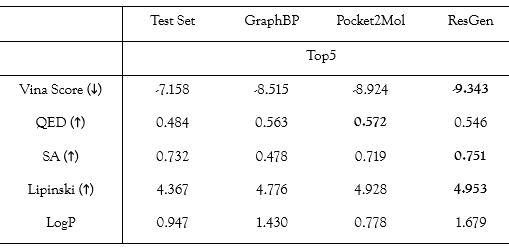
CrossDock 測試集上的 Top5 分子性質
對比結果顯示,ResGen 生成的分子在包括結合能和藥物相似性在內的大部分指標上都優于 GraphBP 和 Pocket2Mol 生成的分子。
* GraphBP:采用 3D 圖神經網絡來提取語義信息,然后通過自回歸流模型依次生成原子。通過將特定類型和位置的原子逐個放置到給定的結合位點來生成與給定蛋白質結合的 3D 分子。
* Pocket2Mol:用于建模三維蛋白質口袋的化學和幾何特征,并采用一種新的高效算法來采樣基于口袋條件的新的3D候選藥物。
如上圖所示,Vina Score 代表了生成分子和對應蛋白靶標的結合能,該指標能夠在一定程度上反映模型是否感知到了口袋內的化學環境。
ResGen 在 Vina Score 上的表現意味著,ResGen 更有機會生成和靶標結合更緊密的分子,研究團隊認為這可能是因為 ResGen 采用了多尺度建模表征結構,因為這種結構更有利于捕捉蛋白質口袋和配體間更高層次的相互作用(如片段-殘基相互作用)。
此外,能否將一個有機化合物推進為候選藥物,不僅取決于其與蛋白質相互作用的強度,還取決于它的類藥性和可合成性。因此 QED、SA、Lipinski 以及 LogP 這些類藥性指標被納入評估。ResGen 在 SA 和 Lipinski 指標上得分最高,表明 ResGen 更有可能為未認知的蛋白質口袋生成易于合成的類藥配體。
針對真實靶標的分子生成:評估現實場景中的表現
為了對模型在真實藥物設計場景中的表現進行評估,研究團隊以蛋白激酶 B 中 AKT1 和 CDK2 (Cyclin-Dependent Kinase 2) 作為案例,整理了其靶標結構以及具有實驗活性的配體化合物,并隨機選擇了一批無活性小分子作為陰性對照。
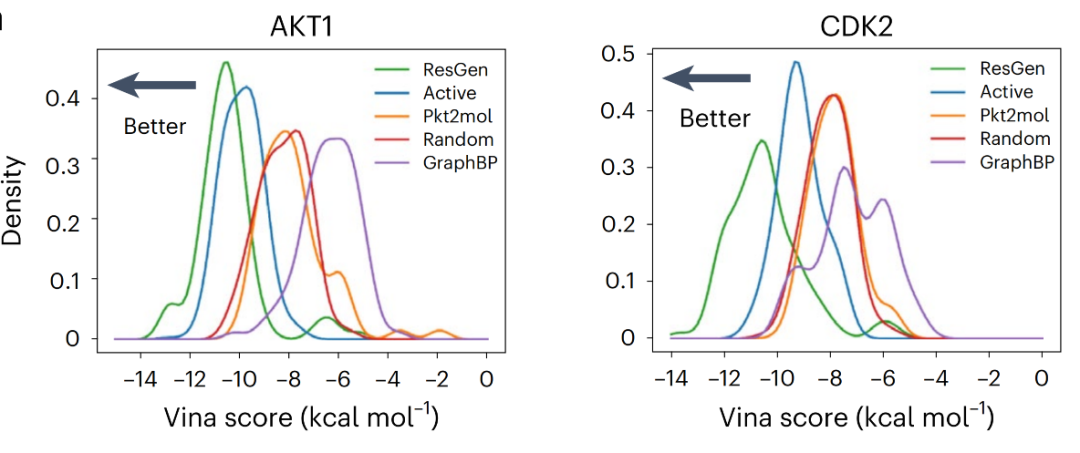
上圖展示了各組分子的結合親和力分布,分布越偏左,結合能絕對值越大,親和力越高。結果表明,ResGen(綠色) 生成的分子不僅比陰性對照 (Random) 和其他現有最先進模型生成的分子得分更高,而且整體分布甚至略好于 Active。
鍵長分布實驗:評估構象合理性
在構象合理性實驗中,研究團隊計算了直接生成的分子構象,與傳統構像軟件生成的分子構象之間的均方根偏差,并比較了生成樣本與訓練分子之間的鍵長分布。
在 7 種鍵長中,ResGen 在 5 種鍵長中表現最佳,大大優于 GraphBP(大約 10 倍)。與其他兩個現有的最先進模型相比,ResGen 能生成更平滑的構象,這突出了其在捕捉蛋白質口袋內部復雜幾何分布方面的強大能力。
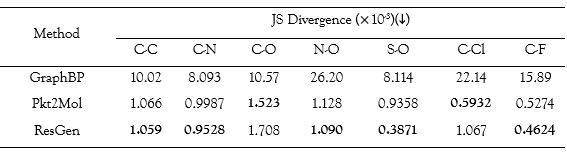
不同方法的鍵長分布與訓練集的鍵長分布比較
AlphaFold 預測結構分析:評估模型對相互作用的敏感度
為驗證 ResGen 是否成功學習了依賴靶點幾何結構的相互作用模式,以及模型對蛋白-小分子相互作用的敏感度,研究團隊以 X 射線晶體結構和 AlphaFold 預測結構為條件分別生成了兩組分子,并對了比這兩組分子的結構特征。

基于晶體結構和 AlphaFold 預測結構生成的分子。其中白色配體為共晶配體,X ? 為經過對齊后預測結構與真實結構間的 RMSD。第一列中的白色圓球代表可能的結合位點。
AlphaFold 預測的構象 「封閉 」了晶體構象中存在的口袋,導致模型無法在原口袋位置生成完整的分子,而是在新形成的空腔中生成小片段,表明了 ResGen 的分子生成過程靈敏地依賴于給定的蛋白質口袋。

AlphaFold 預測構象中形成的口袋與晶體口袋相比差異較小,但是模型仍然可以捕捉到這種變化。ResGen 生成的分子更多地占據了 AlphaFold 預測構象中的空腔結構(如圖中紅圈所示)。
這一實驗證明了 ResGen 對靶點結構的敏感性,也暗示了正確的蛋白結構對于 SBMG 策略的重要性。
「AlphaFold2 推理蛋白質結構」詳細教程:
https://openbayes.com/console/public/tutorials/m6k2bdSu30C
AlphaFold 蛋白質結構數據集:
https://openbayes.com/console/public/datasets/ETTgyY1oZat/1/overview
點擊「閱讀原文」即可一鍵 input,無需下載數據集
侯廷軍:致力于計算機輔助藥物設計核心問題的研究
分子生成是一項典型的多目標優化任務。我們生成的分子不僅希望他具有好的親和力,還需要有好的成藥性,低的毒性,高的合成性等。
——侯廷軍
在傳統的藥物發現過程中,藥物創新存在研發周期長、投入高、風險大等問題。先導化合物的發現和優化是整個藥物發現過程中最具挑戰性的階段,需要克服化合物化學空間巨大的難題(可能達到 10 的 60 次方量級);此外,先導物的篩選、優化和評價過程非常復雜。
而通過深度學習和大數據分析,AI 能夠高效處理和解讀大規模的生物信息學數據,挖掘隱藏在龐大數據集中的模式和關聯,提高對潛在藥物靶點的識別準確性,加速藥物篩選和設計的過程。
面向 AI 輔助藥物研發領域,侯廷軍教授及其團隊長期圍繞計算機輔助藥物設計中的核心問題,展開前沿交叉學科研究,并取得一系列頗具價值的成果,例如:
* 分子對接虛擬篩選方面,提出新型的基于圖表征學習的蛋白-小分子相互作用的打分方法 IGN、基于深度學習的高通量分子對接框架 KarmaDock 等。
*智能分子生成和優化方面,提出基于配體的多約束分子生成方法 MCMG、基于拓撲表面和幾何結構的 3D 分子生成方法 SurfGen 等。
*分子成藥性及安全性評估方面,提出基于多圖注意力模型的毒性預測方法 MGA,成藥性預測軟件系統 ADMETlab2.0 等。
除此之外,侯廷軍教授團隊還研發了基于子結構掩蓋的 AI 模型可解釋性方法 SME,對 AI 模型的可解釋性提出了解決方法。
盡管 AI 在藥物研發中發揮的巨大價值日益凸顯,但作為新興研究,在實際落地中或許還存在相應的挑戰,而這些恰恰也將成為未來的重點研究方向。
對此,侯廷軍教授表示,如何有效提升基于 AI 的性質預測方法的預測能力、基于 AI 的打分函數在虛擬篩選中的預測能力、關鍵成藥性參數和毒性終點的預測精度,將是 AI 輔助藥物發現領域未來需要重點關注的方向與挑戰。
參考資料:https://mp.weixin.qq.com/s/cxpbeGmrHULcWsbVbvQmJA
來源: HyperAI超神經
