
 科普中國公眾號
科普中國公眾號
 科普中國微博
科普中國微博

 幫助
幫助
 化學加
化學加 
導讀
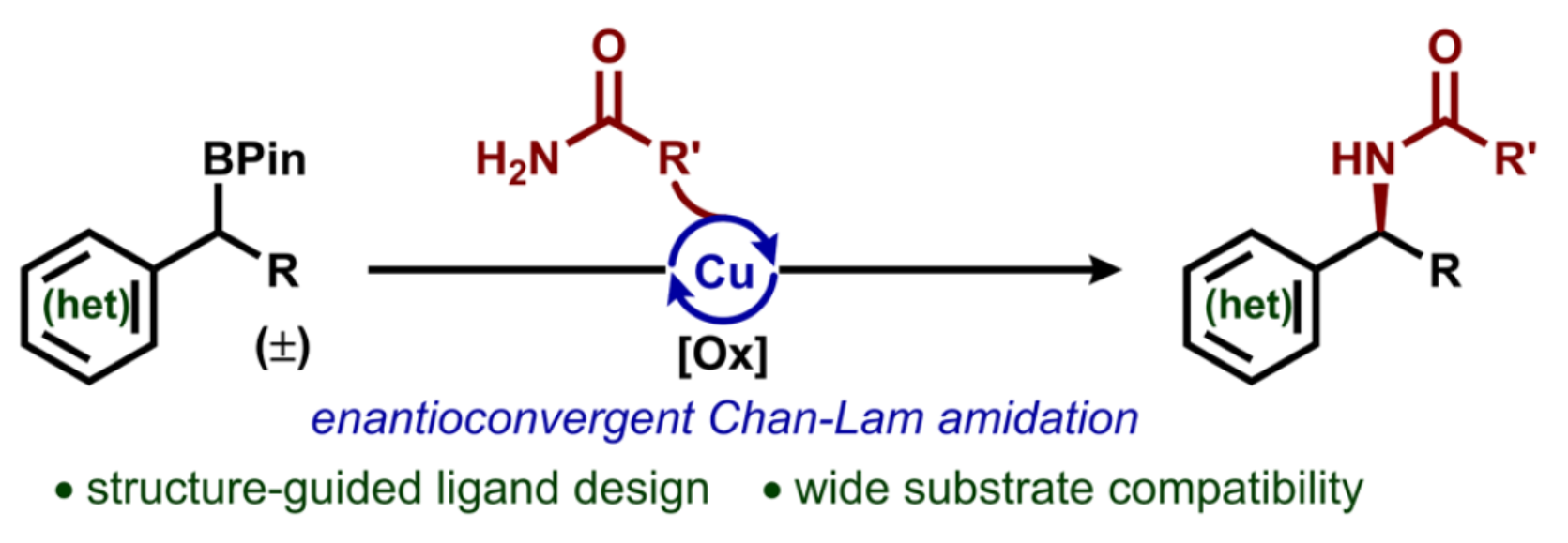
近日,美國科羅拉多州大學Robert S. Paton與Yuyang Dong團隊報道了一種采用模塊化的烷基頻哪醇硼酸酯為底物,首次實現了對映匯聚性Chan?LamC(sp3)?N偶聯反應。該策略具有高度的對映選擇性,可耐受多種官能團、雜環結構及藥物相關分子骨架。其次,通過Hammett分析、自由基鐘以及自由基捕獲實驗,進一步研究了反應的機理。此外,DFT計算支持自由基接力反應路徑:C?B鍵發生氧化均裂生成前手性烷基自由基,該自由基通過內球層機理被原位生成的Cu(II)中間體官能團化。文章鏈接DOI:10.1021/jacs.5c05884
(圖片來源:J. Am. Chem. Soc.)
正文
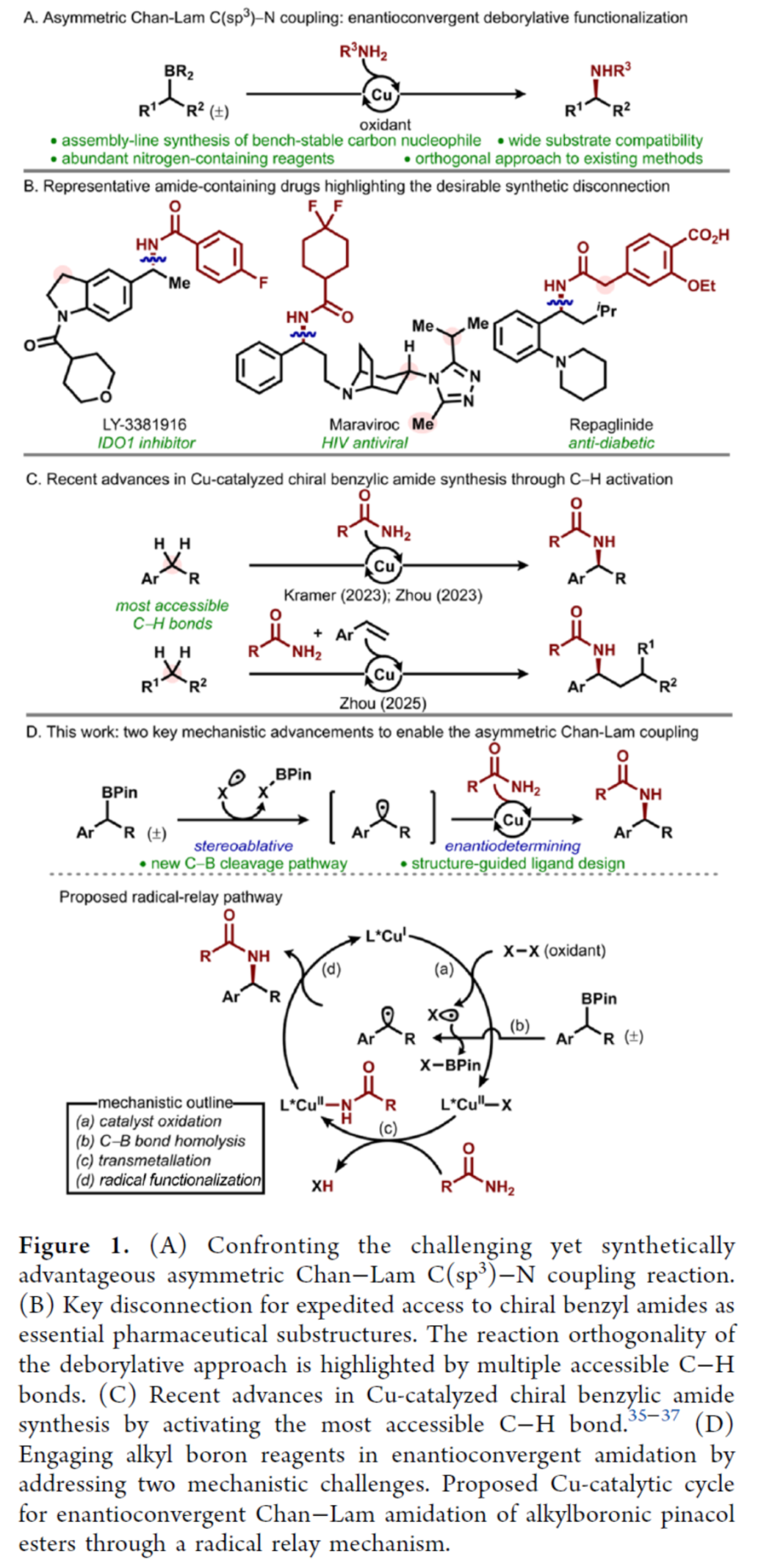
有機硼試劑的氧化Chan?Lam胺化反應是一種合成藥物活性成分的高效交叉偶聯策略。盡管Chan?Lam偶聯反應在芳胺合成中應用廣泛,但其在對映選擇性構建C(sp3)?N鍵領域的應用仍存在局限。這一挑戰源于通常依賴于使用超化學計量量的銅試劑,且缺乏適用于烷基硼試劑參與的普適性機理框架。然而,這一轉化反應通過結合Chan?Lam偶聯的普適性與烷基硼試劑的合成多功能性,能夠實現強大的逆合成斷鍵策略,從而構建復雜烷基骨架(Figure 1A)。作者認為,烷基硼酸頻哪醇酯和酰胺之間的脫硼交叉偶聯反應將為獲得手性芐基酰胺提供一種有利的策略,其是一類特殊的藥物亞結構(Figure 1B)。該策略不僅為傳統依賴手性助劑或拆分工藝的多步合成法提供了簡化且模塊化的替代路徑,同時可作為新興催化策略(如近期C?H鍵官能團化策略)的正交互補方案(Figure 1C)。作者設想,通過解決兩大關鍵機理挑戰可達成該轉化:(1)烷基頻哪醇硼酸酯中C?B鍵的均裂;(2)經由銅(II)酰胺中間體實現烷基自由基的對映選擇性官能團化(Figure 1D)。具體反應過程如下:通過自由基接力路徑(step a)形成的高活性有機自由基,可應用于類似反應過程(step b)。所產生的前手性烷基自由基將與原位生成的Cu(II)酰胺中間體反應,進而生成光學活性產物(step c-d)。在此,美國科羅拉多州大學Robert S. Paton與Yuyang Dong團隊首次報道了一種通過配體設計促進的對映匯聚性Chan?LamC(sp3)?N偶聯反應,涉及自由基介導的脫硼官能團化的過程(Figure 1D)。歡迎下載化學加APP到手機桌面,合成化學產業資源聚合服務平臺。
(圖片來源:J. Am. Chem. Soc.)
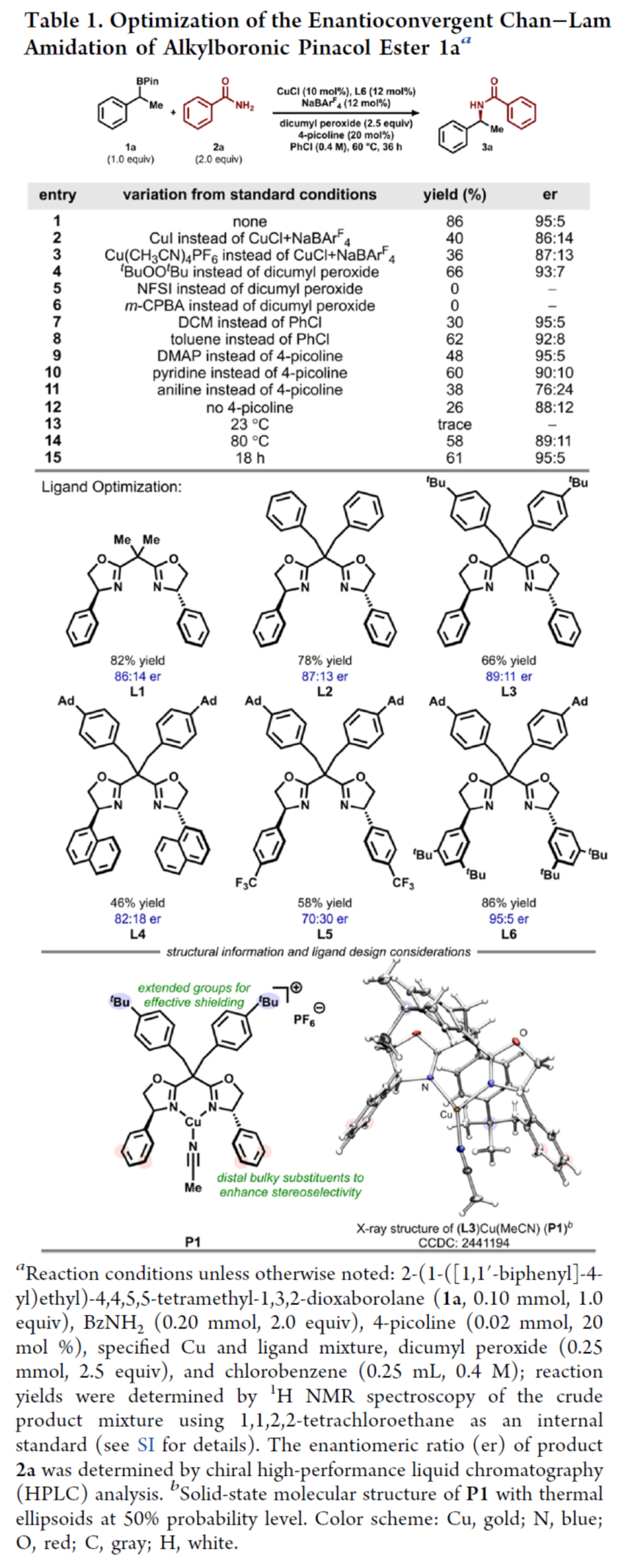
首先,作者以烷基硼酸頻哪醇酯1a與苯甲酰胺2a作為模型底物,進行了相關反應條件的篩選(Table 1)。當以CuCl(10 mol %)作為催化劑,NaBArF4(12 mol %)作為添加劑,L6(12 mol %)作為配體,4-甲基吡啶(20 mol %)作為Lewis堿性添加劑,過氧化二異丙苯(2.5 equiv)作為氧化劑,在PhCl溶劑(0.4 M)中60 oC反應36 h,可以86%的收率得到產物3a,er為95:5。
(圖片來源:J. Am. Chem. Soc.)
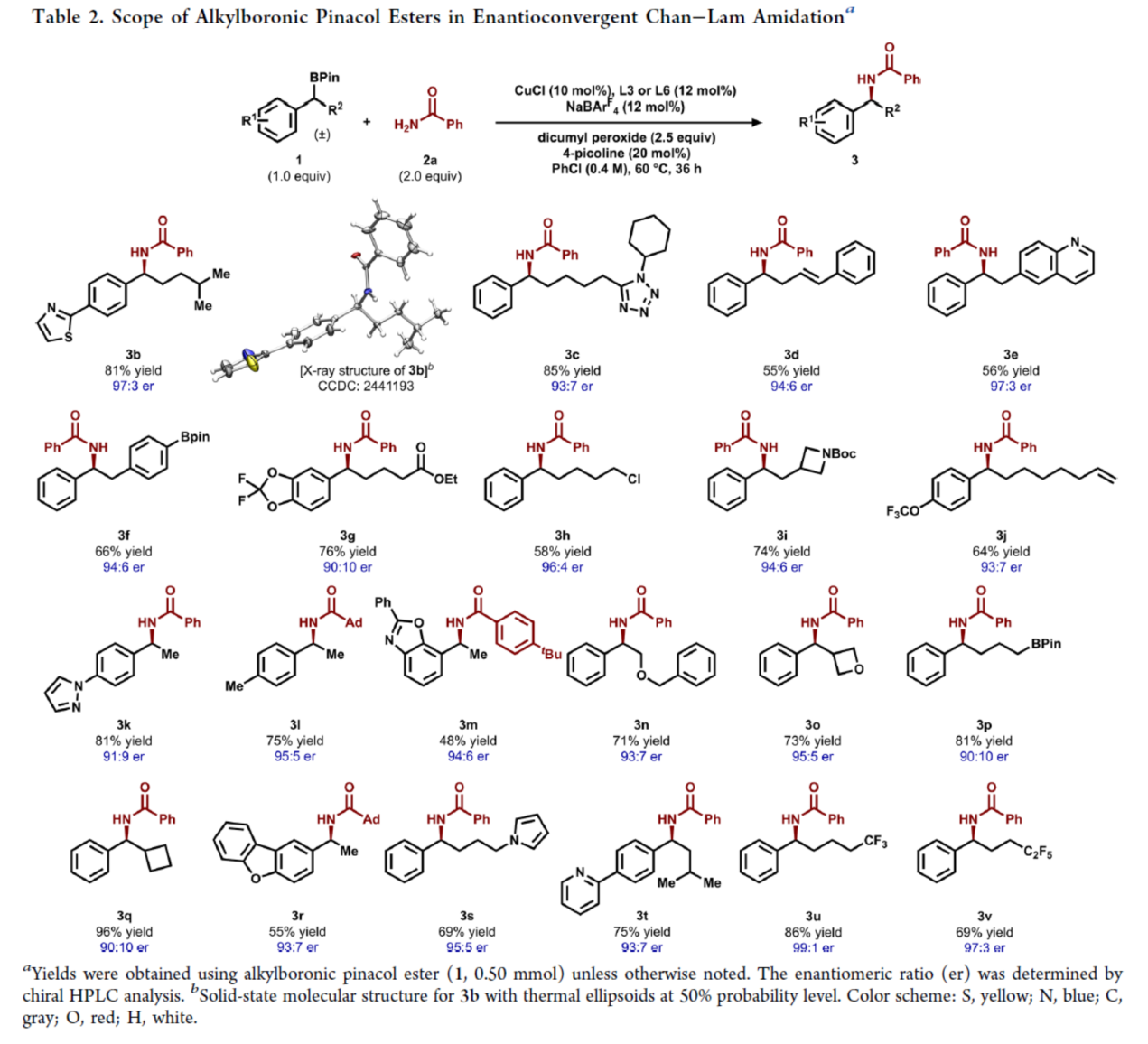
在獲得上述最佳反應條件后,作者對烷基硼酸頻哪醇酯的底物范圍進行了擴展(Table 2)。研究結果表明,當烷基硼酸頻哪醇酯底物1中含有不同電性取代的芳基、雜芳基或者含有各種烷基與環烷基取代時,均可順利反應,獲得相應的產物3b-3v,收率為48-96%,er為90:10-99:1。
(圖片來源:J. Am. Chem. Soc.)
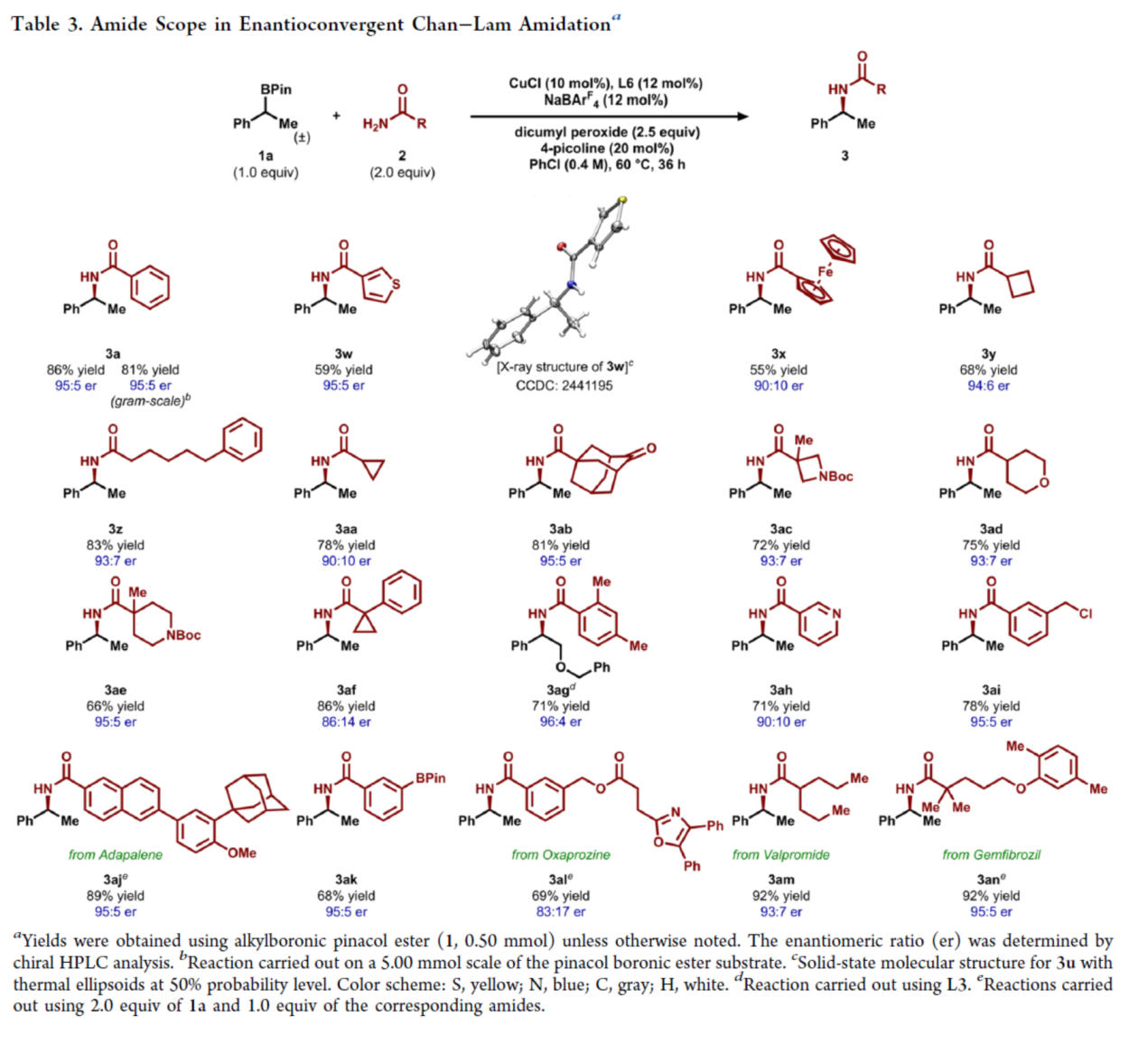
緊接著,作者酰胺的底物范圍進行了擴展(Table 3)。研究結果表明,當酰胺底物2中含有不同取代的芳基、雜芳基、烷基與環烷基時,均可順利反應,獲得相應的產物3a、3w-3z、3ak和3aa-3ai,收率為55-86%,er為86:14-96:4。值得注意的是,該策略還可用于多種藥物分子的后期衍生化,如維生素A衍生物阿達帕林(3aj)、非甾體抗炎藥奧沙普秦(3al)、抗癲癇藥丙戊酰胺(3am)以及降脂劑吉非羅齊(3an),收率為69-92%,er為83:17-95:5。
(圖片來源:J. Am. Chem. Soc.)
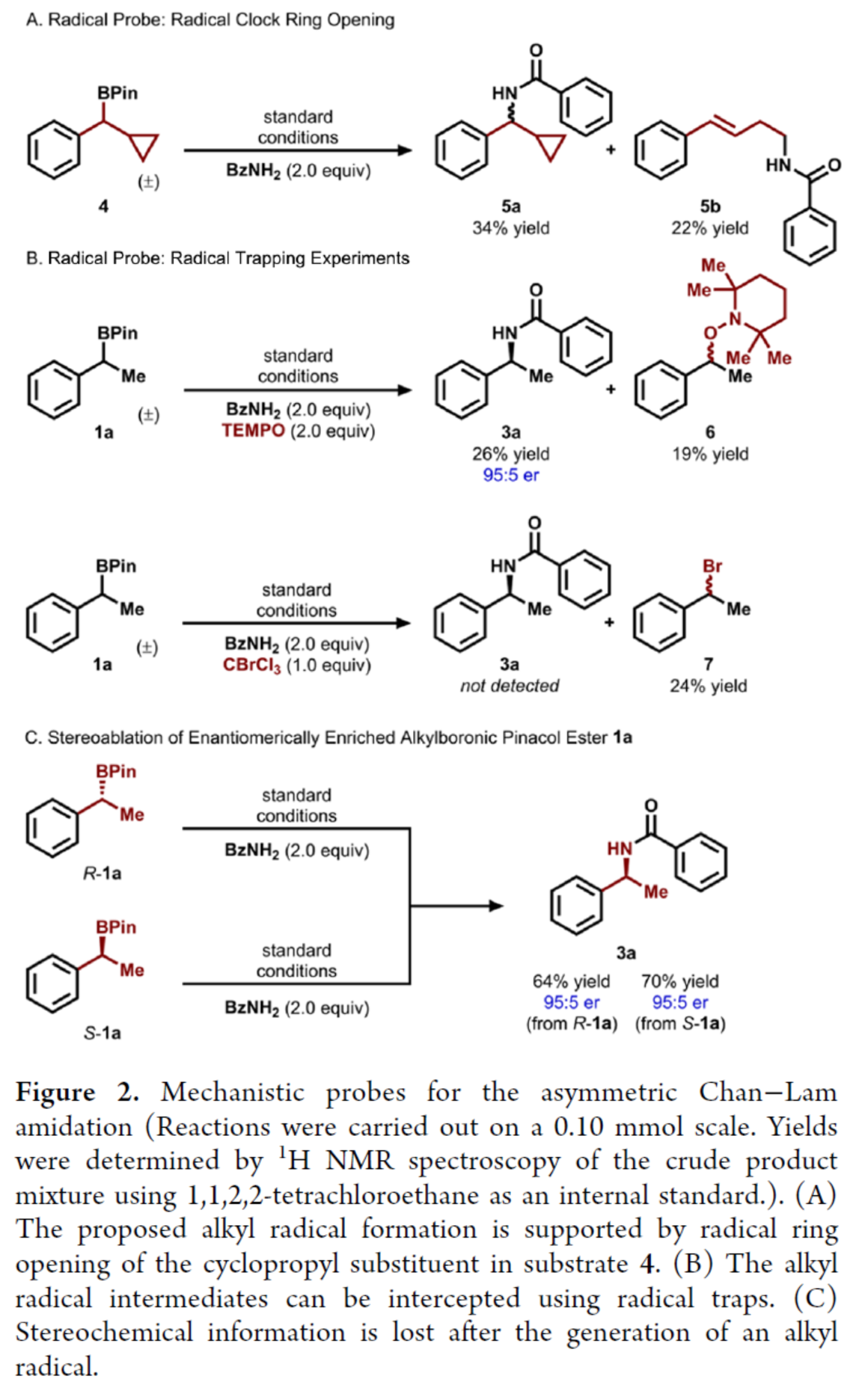
隨后,作者對反應的機理進行了研究(Figure 2)。首先,自由基鐘實驗(Figure 2A)與自由基捕獲實驗(Figure 2B)結果表明,反應涉及烷基自由基的生成。其次,將對映體富集的(R)-1a和(S)-1a置于標準條件下,均生成具有相似對映體比例的產物3a,表明了在C-N鍵形成之前已發生立體化學信息損失(Figure 2C)。
(圖片來源:J. Am. Chem. Soc.)
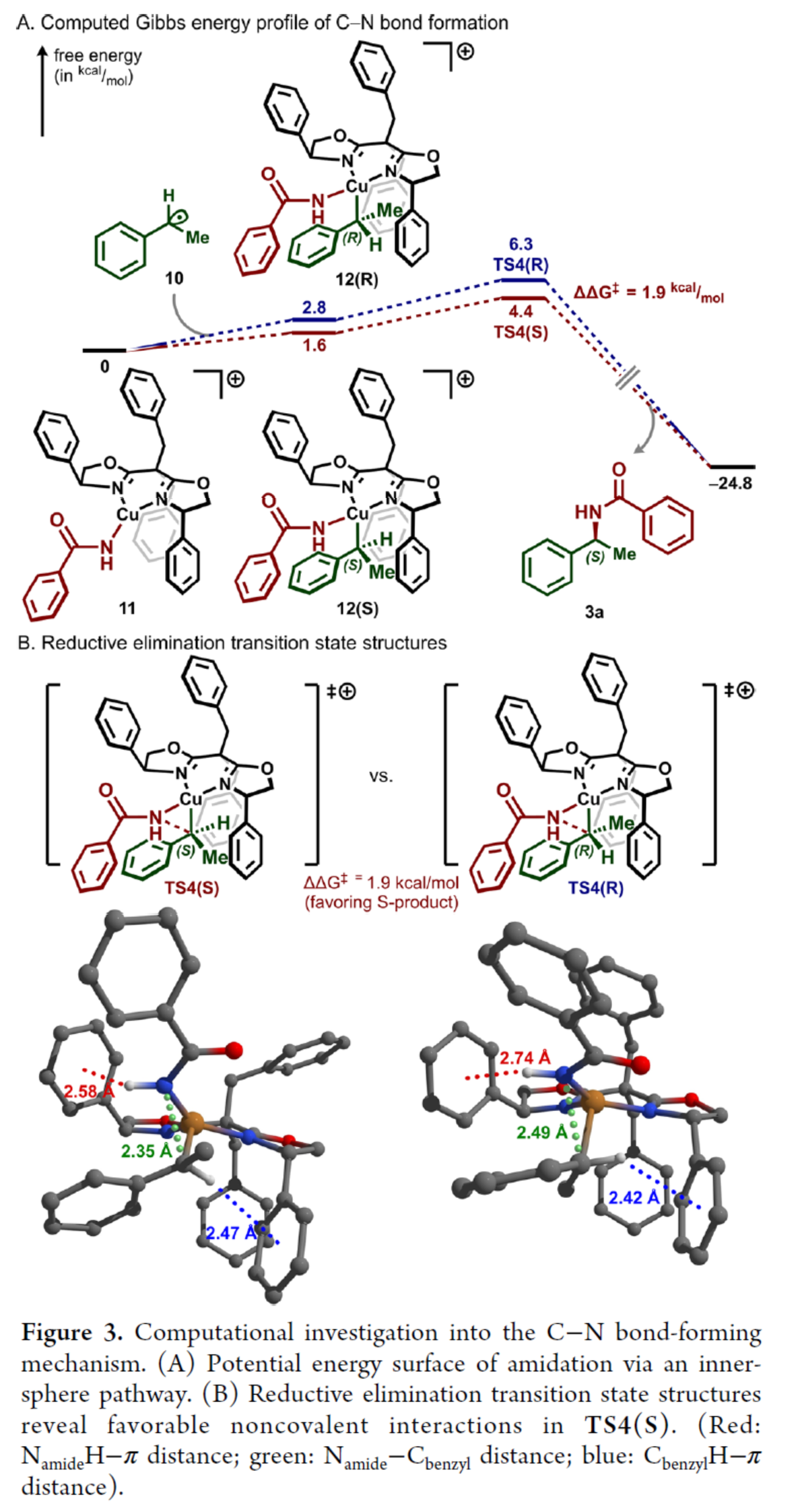
隨后,作者通過相關的理論計算,探究了C-N鍵形成機理并闡明了其對映匯聚性的起源(Figure 3)。計算結果表明:二價銅中間體11的形成是放熱過程,其反應路徑為先經二異丙苯過氧化物氧化,再與苯甲酰胺2a發生金屬轉移反應(Figure 3A)。烷基自由基10和二價銅中間體11的結合具有輕微的吸電子性,形成形式的Cu(III)中間體12(R)和12(S)。隨后,C-N鍵形成過程通過還原消除過渡態結構TS4(R)和TS4(S)進行,且芐位構型保持。其中,計算結果表明,反應有利于S-對映異構體的形成(Figure 3B)。同時,S-對映體的優勢源于TS4(S)中更強的NamideH?π相互作用。
(圖片來源:J. Am. Chem. Soc.)
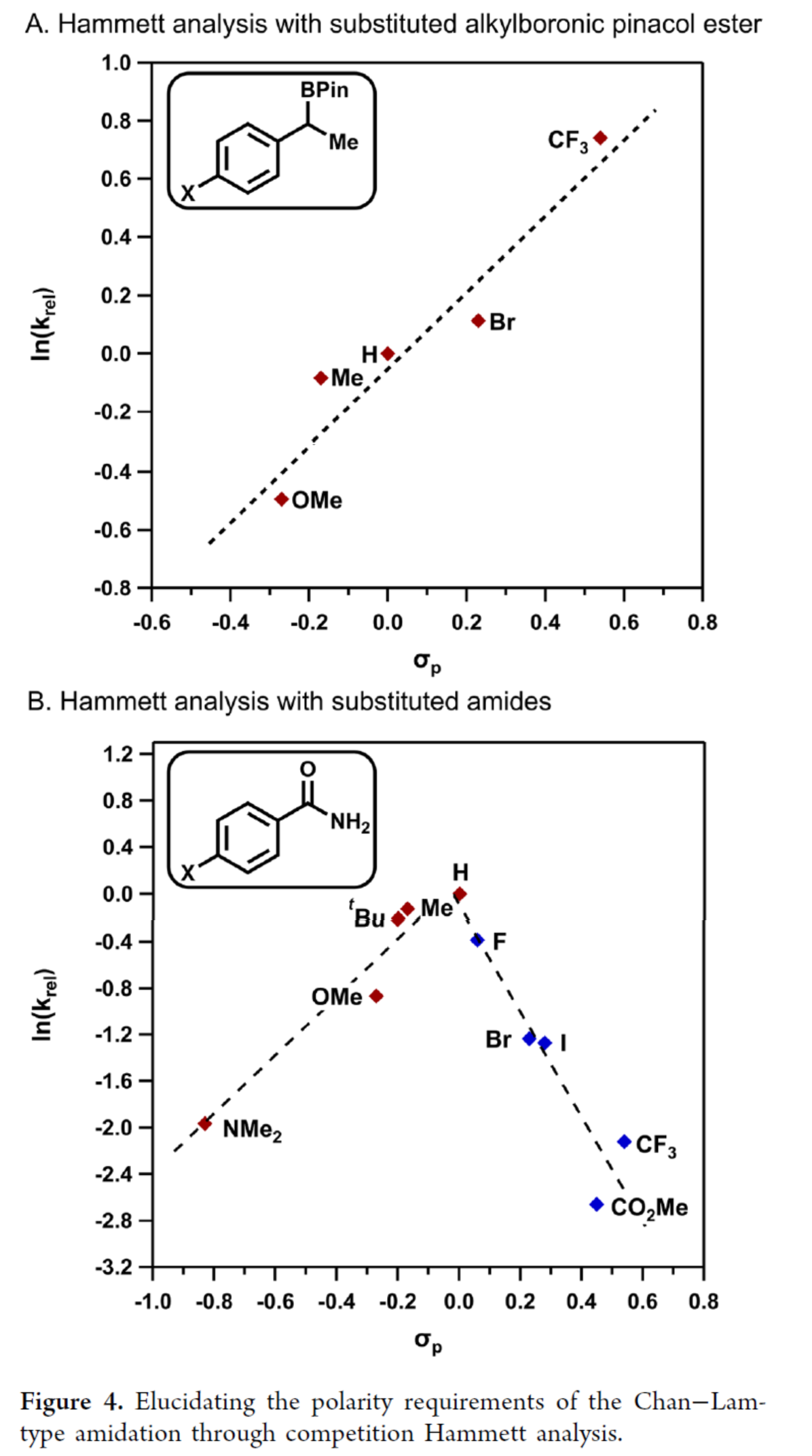
最后,作者通過競爭性Hammett分析闡明Chan-Lam型酰胺化的電子效應需求(Figure 4)。通過分析取代烷基硼酸頻哪醇酯的Hammett參數與反應速率的相關性,揭示出正斜率趨勢(Figure 4A)。該現象可歸因于:(1)C-B鍵斷裂前,枯氧自由基?8?與親電性更強的芐基硼酸酯之間存在有利結合;(2)親電性更強的芐基自由基提升了C-N偶聯效率。相應地,缺電子酰胺的反應速率低于苯甲酰胺(Figure 4B),這很可能源于極性失配導致。然而,作者也觀察到富電子酰胺反應速率減緩,導致Hammett相關曲線斜率發生變化。向下凹陷的Hammett圖表明限速步驟發生轉變。作者認為,富電子酰胺的較低酸性導致轉金屬化過程變慢。基于上述機理框架,作者正在進行不對稱Chan-Lam型交叉偶聯實驗方案的開發,以適應廣泛的親核試劑。
(圖片來源:J. Am. Chem. Soc.)
總結
美國科羅拉多州大學Robert S. Paton與Yuyang Dong團隊開發了首個不對稱Chan?LamC(sp3)?N偶聯反應。該策略對各類官能團及雜環結構均具有良好兼容性,可在克級規模上以高度的對映選擇性獲得相應的酰胺化產物。實驗與理論計算研究共同支持其經由內球層C-N偶聯機理的自由基接力反應路徑,其中立體選擇性源于配體-底物間的非共價相互作用。
文獻詳情:
Enantioconvergent Chan?Lam Coupling: Synthesis of Chiral Benzylic Amides via Cu-Catalyzed Deborylative Amidation.
Jonathan Vu, Graham C. Haug, Tanner J. Schubert, Joshua F. Head, Robert S. Paton,*Yuyang Dong*.
J. Am. Chem. Soc.2025
DOI:10.1021/jacs.5c05884
來源: 化學加
